l’Hémoglobinurie Paroxystique Nocturne
Hémoglobinurie, caractérisant la présence d'hémoglobine libérée par la destruction des globules rouges dans les urines.2
Paroxystique, pour les poussées soudaines 2
Nocturne, car survenant la nuit 2
Zoom sur la prise en charge de l'HPN à l'hôpital Saint-Louis, centre de référence de la pathologie
L’hémoglobinurie paroxystique nocturne (HPN) est une maladie rare et grave, impactant profondément la vie des patients. Comment est-elle prise en charge ? Quels sont les défis pour les professionnels de santé et les patients ?
Dans cette vidéo immersive, découvrez le rôle clé du Centre de Référence des aplasies médullaires et HPN à l’Hôpital Saint-Louis, grâce aux témoignages de :
- Pr Régis Peffault de Latour, hématologue et coordonnateur du centre ;
- Dr Flore Sicre de Fontbrune, hématologue ;
- Sylvia Derouet, infirmière en hôpital de jour.
Mieux comprendre
L’Hémoglobinurie Paroxystique
Nocturne
L’HPN c’est quoi ?
L’hémoglobinurie paroxystique nocturne (HPN) est une maladie hématologique qui se caractérise par un phénomène de destruction prématurée de globules rouges connu sous le nom d’hémolyse.1
L’HPN touche 600 personnes en France. C’est une maladie chronique rare dont la présentation clinique et la gravité varient, pouvant retarder ou compliquer son diagnostic. L’errance diagnostic pour ces patients peut aller jusqu’à 5 ans. Potentiellement mortelle, les personnes atteintes d’HPN non diagnostiquées et non traitées risquent de mourir d’une thrombose, d’une hémorragie ou d’une infection. En effet, en l'absence de traitement 20 à 40% des patients atteints d'HPN décèdent dans les 5 à 6 ans suivant le diagnostic. 3,4,5,6
Quelle est l’origine de la maladie ?
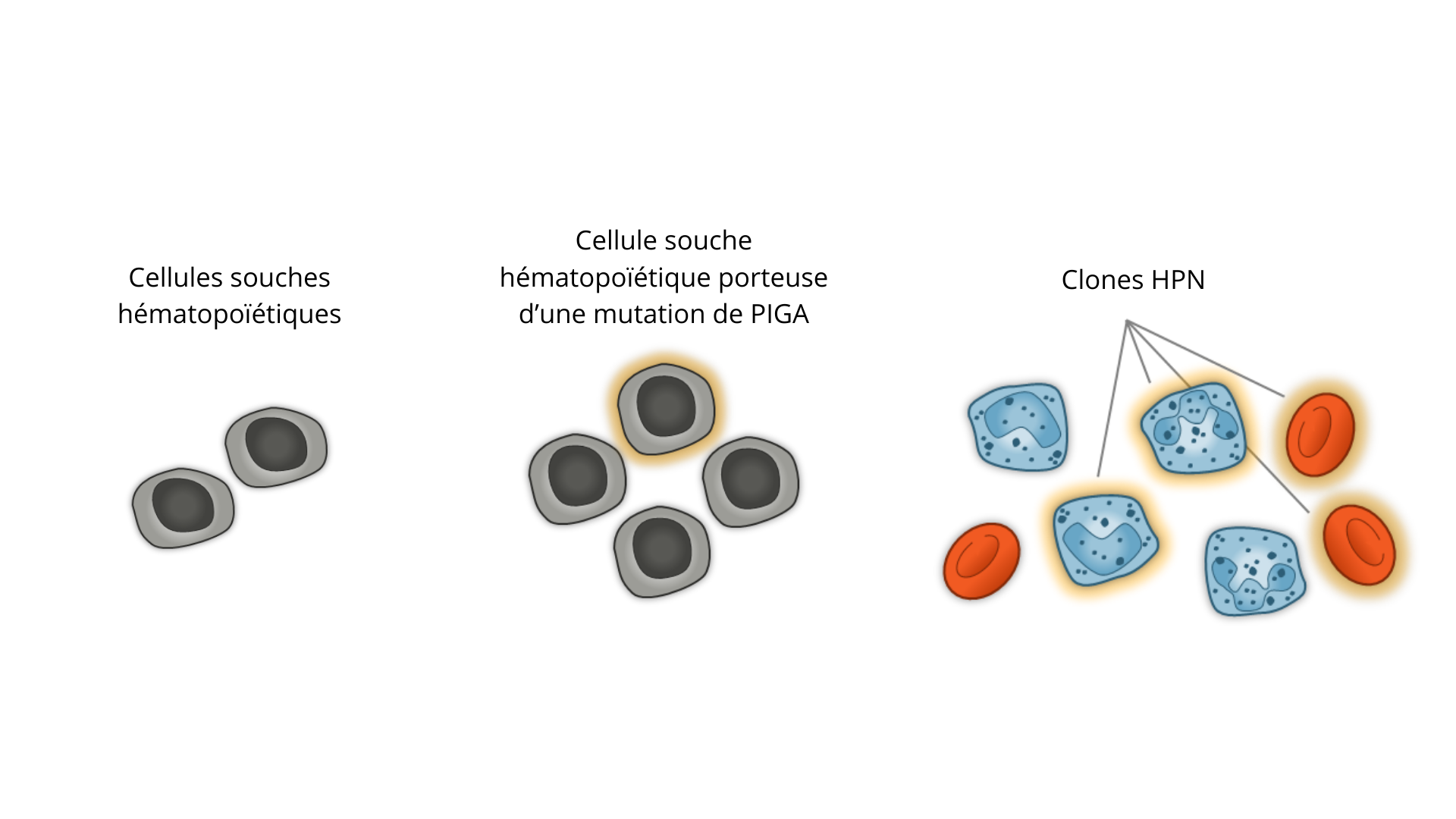
L’hémoglobinurie paroxystique nocturne survient lorsque les cellules souches hématopoïétiques (CSH) responsables de la fabrication des cellules sanguines de la moelle osseuse, mutent et commencent à produire des cellules sanguines défectueuses ayant perdues l’expression à leur surface de certaines protéines. 1,2,7
La maladie résulte d'un défaut dans le mécanisme de production des globules rouges - plus précisément une mutation dans un gène d'une CSH appelé le gène PIGA. Ces cellules souches porteuses de mutations du gènes PIG-A vont se multiplier et conduire à la formation de cellules sanguines matures porteuses de la même mutation déficitaires en protéines GPI-ancrées. Les CSH mutantes forment alors une population de cellules sanguines mutantes appelées clones HPN. 1,8,9
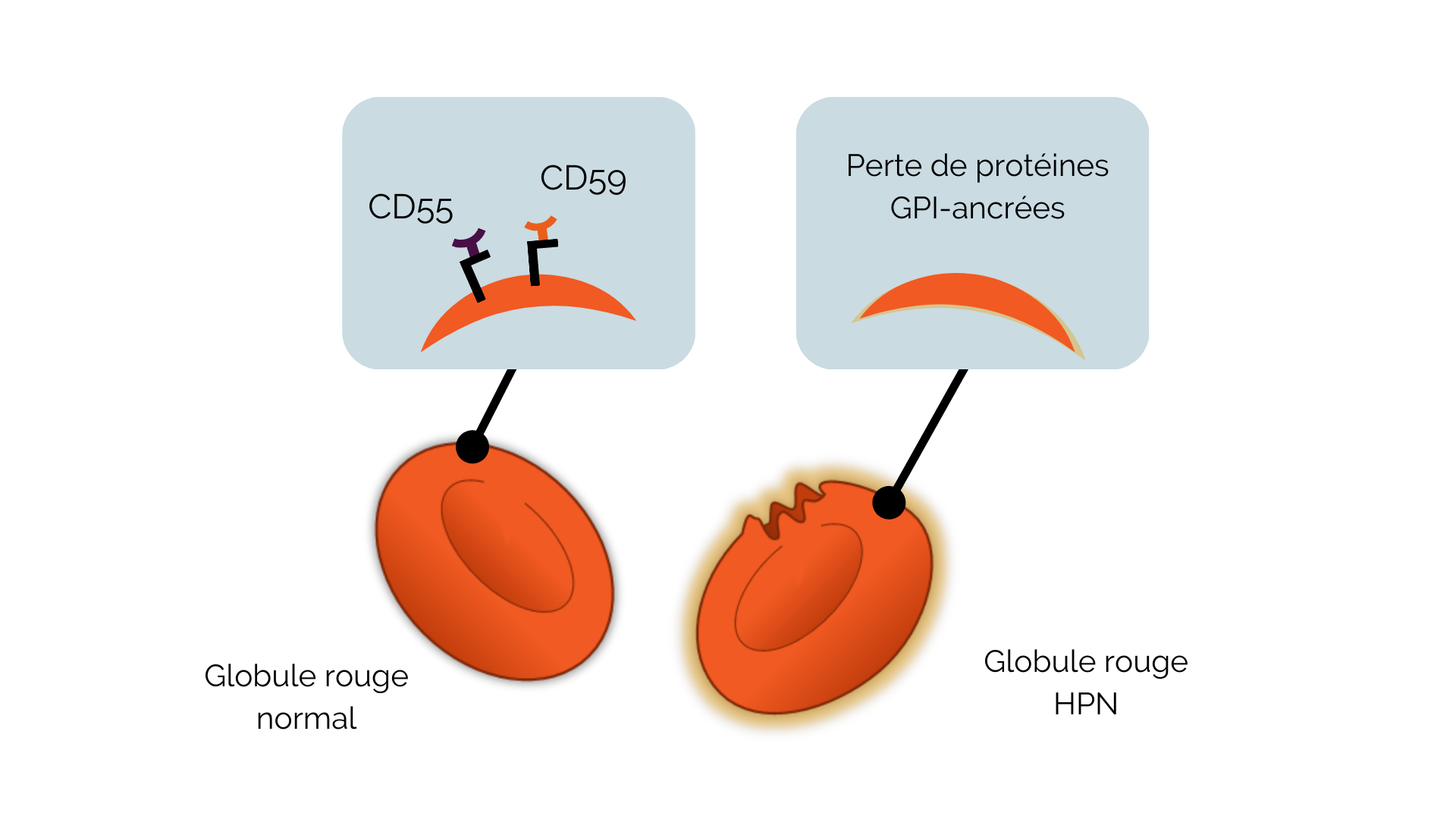
Dans l’HPN, les mutations du gène PIGA entraînent la perte de CD55 et CD59, qui provoque à son tour la destruction des globules rouges médiée par le complément1
Protéines GPI-ancrées : CD55 et CD59
Le gène PIGA est requis pour la synthèse des ancres GPI, et donc pour assurer la présence de protéines spécialisées à la surface des cellules1
CD55 et CD59 sont des types de protéines GPI-ancrées. Ce sont des régulateurs négatifs du complément qui empêchent les cellules de l’organisme d’être attaquées par le système du complément.1
Les manifestations cliniques d’HPN surviennent lorsqu’un clone de CSH porteur de mutations somatiques du gène PIGA acquiert un avantage de croissance et se différencie, créant une proportion importante de cellules sanguines matures déficitaires en protéines GPI-ancrées. Les globules rouges et granulocytes déficitaires en CD55 et CD59 qui en résultent sont appelés collectivement clones HPN. Le pourcentage de globules rouges ou granulocytes présentant un déficit en CD55 et CD59 ce que l’on qualifie de taille du clone HPN. 1,10
Le système du complément est une composante centrale du système immunitaire impliquée dans la défense contre les agents pathogènes, l’homéostase de l’hôte, et la régulation de la réponse immunitaire. Composé de protéines, de convertases, de récepteurs et de régulateurs, le système du complément facilite l’élimination des agents pathogènes nocifs et des cellules de l’organisme qui sont endommagées par des phénomènes d’inflammation, de lyse directe, d’opsonisation et de phagocytose. 11,12,13,14
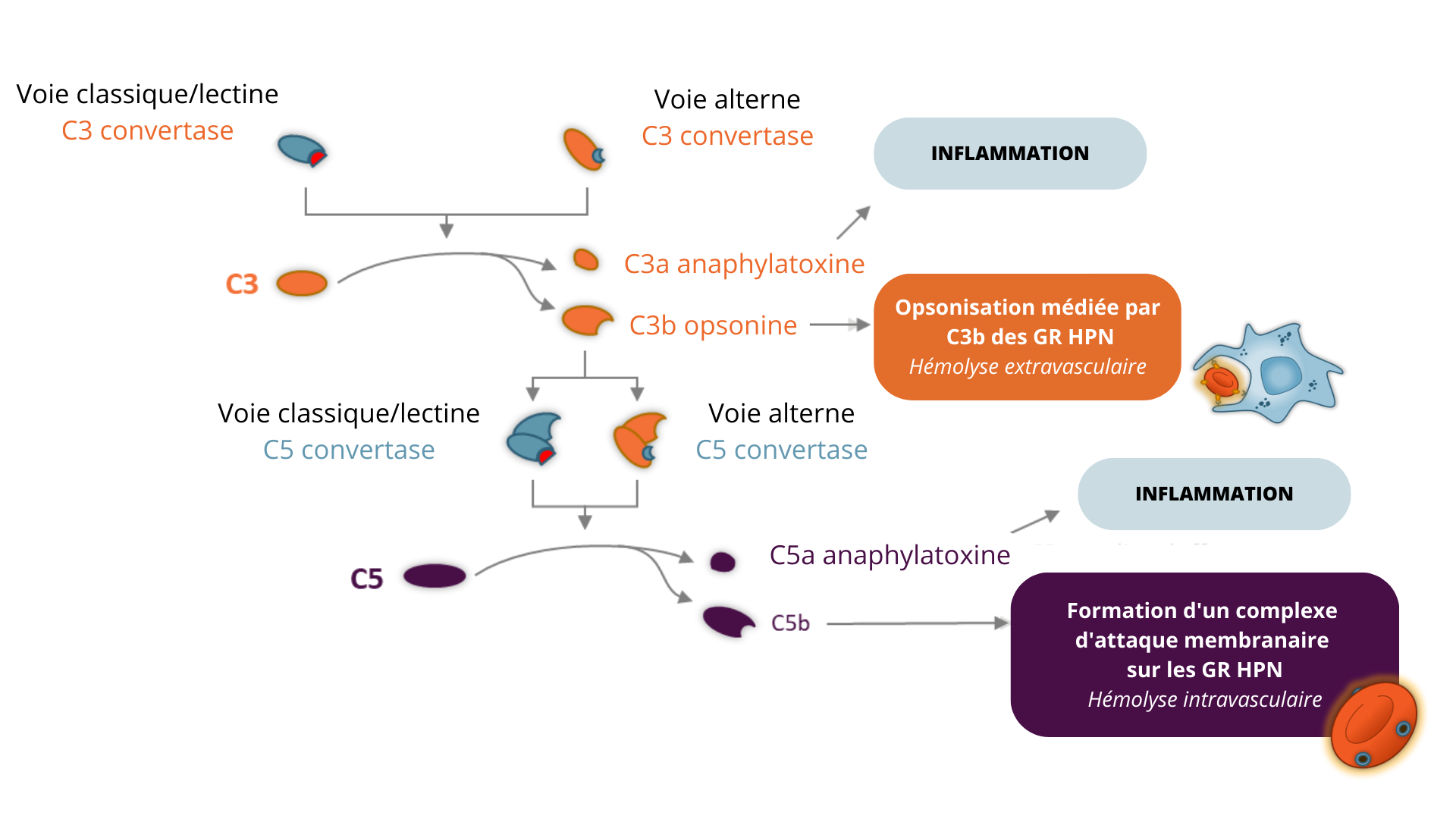
Dans l’HPN, les globules rouges HPN vont être reconnus par le système du complément comme des organismes étrangers en raison de leur modification génétique. L’hémolyse des globules rouges HPN mutant médiée par le système du complément libère dans le plasma l’hémoglobine qu’ils contenaient. Cette libération provoque une anémie modérée à sévère et un ensemble de symptômes et de complications (élévations des taux hémoglobine, thrombose, libération de LDH et de bilirubine). 1,15,16
Il existe deux types hémolyses :
- Hémolyse intra-vasculaire : conséquence de la formation du complexe d’attaque membranaire sur les globules rouges HPN à l’intérieur des vaisseaux sanguins
- Hémolyse extra-vasculaire : résultant de l’opsonisation des globules rouges HPN médiée par C3b qui seront détruits dans le foie et la rate
Tout comprendre sur le rôle de la cascade du complément
Quels impacts pour
le patient ?
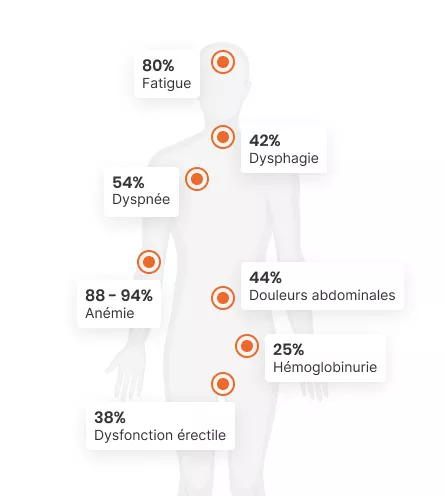
L’HPN est responsable de nombreux signes et symptômes, qui peuvent compliquer sérieusement le quotidien des patients.
De multiples manifestations cliniques chroniques et invalidantes sont souvent présentes dans l’HPN, il peut s’agir notamment de taux d’hémoglobine constamment bas, d’anémie, de fatigue, d’essoufflement, de difficulté à avaler, de maux de tête, d’urine foncée, de douleurs abdominales ou encore de dysfonction érectile.4,5
Des ressources pour mieux en parler à vos patients
Quels sont les traitements disponibles ? 22-27
Traitement curatif 13,14,15
- Greffe de moelle
Traitements spécifiques 16,18
- Les inhibiteurs du complément
Traitement symptomatiques 13,15,16
- Ajustement de dose du traitement spécifique
- Transfusion de globules rouges
- Anticoagulants
- Traitement de l’aplasie associée
- Traitement antibiotique en cas d’infection
Traitement non spécifiques associés 16,17
- Folates : limiter le risque de carence
- Contraception non thrombogène pour les femmes si nécessaires
- Vaccination : limiter le risque d’hémolyse aiguë ; anti- méningococcique ; anti-pneumocoque
Comment définir la réponse au traitement pour les patients HPN ?
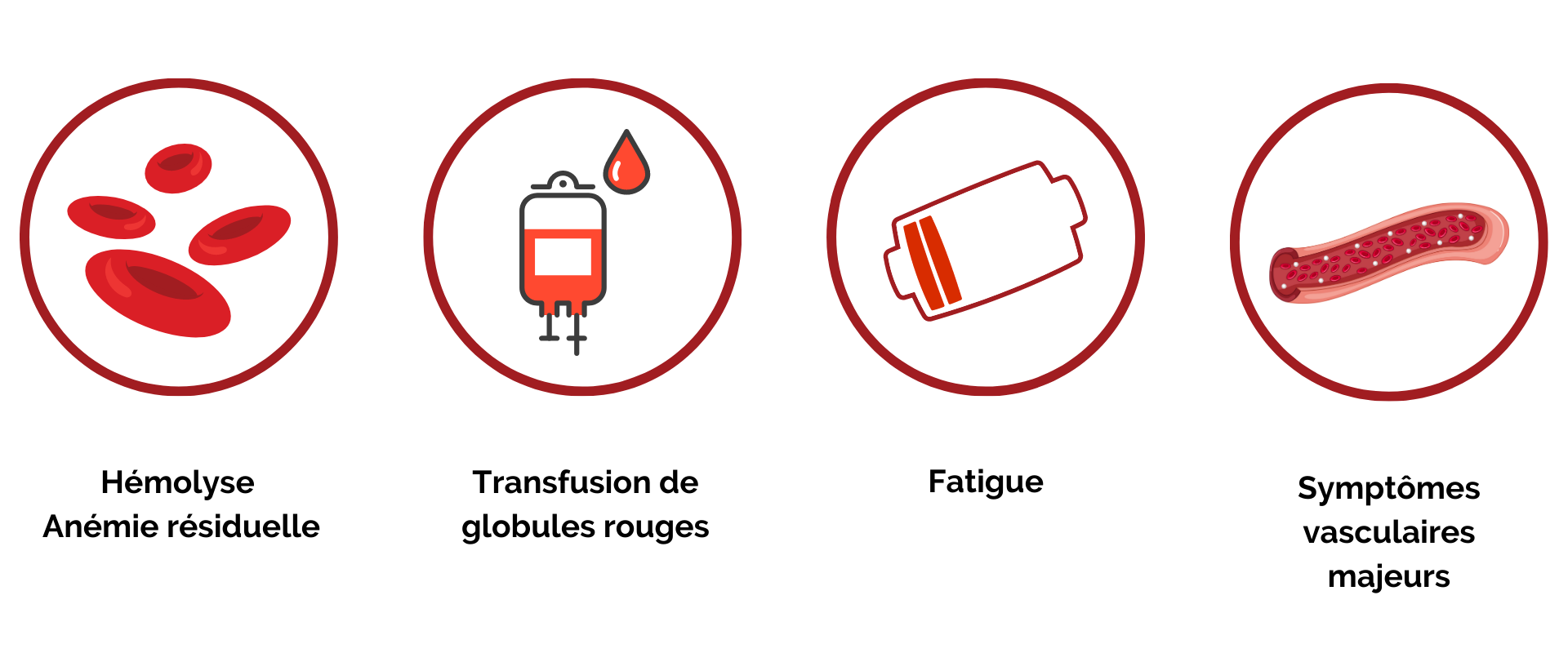
Des symptômes peuvent persister chez les patients HPN sous traitements18-22 :
La réponse au traitement se classe selon 4 catégories qui se définissent comme17 :
- Une réponse complète/majeure : indépendance transfusionnelle et pas d’anémie
- Une bonne réponse : indépendance transfusionnelle et anémie légère (10-12 g/dL)
- Une réponse partielle : anémie persistante (8-10 g/dL) et transfusions occasionnelles (≤ 2
transfusions de globules rouges en 6 mois) - Une réponse mineure / pas de réponse : anémie persistante maintenant les besoins en
transfusions de globules rouges
Pour aller plus loin
dans l’HPN